
About GENIES
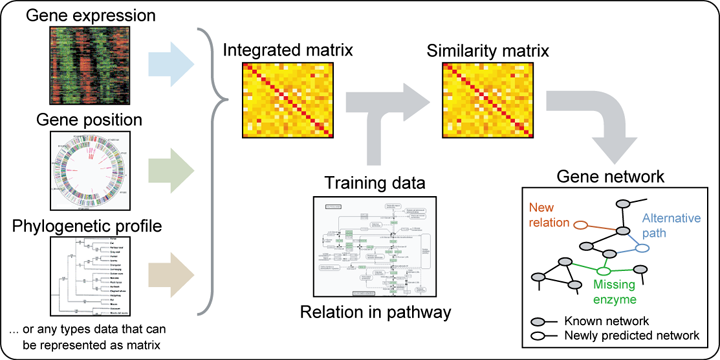
The GENIES (Gene Network Inference Engine based on Supervised Analysis) enables us to predict potential functional associations between genes, based on other genomic data or high-throughput experimental data such as phylogenetic profiles and gene expression profiles.
Usage
The GENIES system has two procedure modes: simple mode and advanced mode. "Simple mode" enables us to conduct a network prediction with default method and parameters. "Advanced mode" enables us to conduct a network prediction with direct or supervised approach with selected parameters.Simple mode
Step 1. Specify an organism in the analysis. Step 2. Upload the data files: tab-delimited text file of "profile" for each dataset (the 1st row corresponds to gene labels and the 1st column corresponds to attributes.) or tab-delimited text file of "kernel matrix" that users have precomputed, (the 1st row corresponds to gene labels and the 1st column corresponds to gene labels.) Step 3. Click on "submit" button.Advanced mode with direct approach
Step 1. Specify an organism in the analysis. Step 2. Upload the data files: tab-delimited text file of "profile" for each dataset (the 1st row corresponds to gene labels and the 1st column corresponds to attributes.) or tab-delimited text file of "kernel matrix" that users have precomputed (the 1st row corresponds to gene labels and the 1st column corresponds to gene labels.) Step 3. Select the method type "direct approach". Step 4. Click on "next" button. Step 5. Assign weight to each dataset in the data integration process. Step 6. Choose kernel type for each dataset if necessary. Step 7. Click on "next" button. Step 8. Put in kernel parameters if necessary. Step 9. Click on "next" button. Step 10. Confirm all the parameters which you have set Step 11. Click on "submit" button.Advanced mode with supervised approach
Step 1. Specify an organism in the analysis. Step 2. Upload the data files: tab-delimited text file of "profile" for each dataset (the 1st row corresponds to gene labels and the 1st column corresponds to attributes.) or tab-delimited text file of "kernel matrix" that users have precomputed (the 1st row corresponds to gene labels and the 1st column corresponds to gene labels.) Step 3. Select the method type "supervised approach". Step 4. Click on "next" button. Step 5. Assign weight to each dataset in the data integration process. Step 6. Choose kernel type for each dataset if necessary. Step 7. Import user's partially known network data. or Use the KEGG pathway data. Step 8. Choose algorithm type for supervised learning ("kernel matrix regression" is encouraged) Step 9. Click on "next" button. Step 10. Put in kernel parameters if necessary. Step 11. Click on "next" button. Step 12. Confirm all the parameters which you have set Step 13. Click on "submit" button.
Output
GENIES has four outputs: 1) Pathway list, 2) Inferred list, 3) Search, and 4) Details.
[ An example of the output page ]
1) Pathway list: predicted interactions related with each pathway
Suppose that you select one pathway map from the pathway list.
The list shows interactions between newly predicted genes and genes belonging to KEGG Pathway.
2) Inferred list: all the predicted gene interaction pairs.
The list shows three kinds of gene interaction pairs categorized into
i) training vs prediction, ii) prediction vs prediction, and iii) training vs training.
3) Search: List of genes which might be related with known enzyme genes.
This option is useful for identifying missing enzyme genes.
Suppose that you select one pathway map and you have a missing enzyme on the pathway.
If you select enzyme boxes in the downside map and click on neighboring enzymes of the missing enzyme,
you will be given a list of newly predicted genes which are associated with known enzymes in the pathway.
4) Details: Change of the parameters and update the result
You can change various parameter values and thresholds and update the results with new parameters.
Data file examples (S. cerevisiae)
1) Gene expression profile (544 genes x 157 experiments)
[ genies_yeast_expmat.txt ]
2) Protein localization data (544 genes x 32 localizations)
[ genies_yeast_locmat.txt ]
3) Phylogenetic profile (544 genes x 145 organisms)
[ genies_yeast_phymat.txt ]
4) Kernel matrix based on the gene expression profile (544 genes x 544 genes)
[ genies_yeast_exp_kernel.txt ]
5) Kernel matrix based on the protein localization profile (544 genes x 544 genes)
[ genies_yeast_loc_kernel.txt ]
6) Kernel matrix based on the phylogenetic profile (544 genes x 544 genes)
[ genies_yeast_phy_kernel.txt ]
Training data for supervised approach
1) Adjacency matrix of partially known network (140 genes x 140 genes)
[ genies_yeast_admat_path.txt ]
Reference
Kotera M, Yamanishi Y, Moriya Y, Kanehisa M, Goto S.; GENIES: gene network inference engine based on supervised analysis. Nucleic Acids Res. (2012) 40 (W1): W162-W167. [NAR] [PubMed]
Yamanishi Y, Vert JP, Kanehisa M.; Supervised enzyme network inference from the integration of genomic data and chemical information. Bioinformatics. (2005) 21 (supple 1): i468-i477. [Bioinformatics] [PubMed]