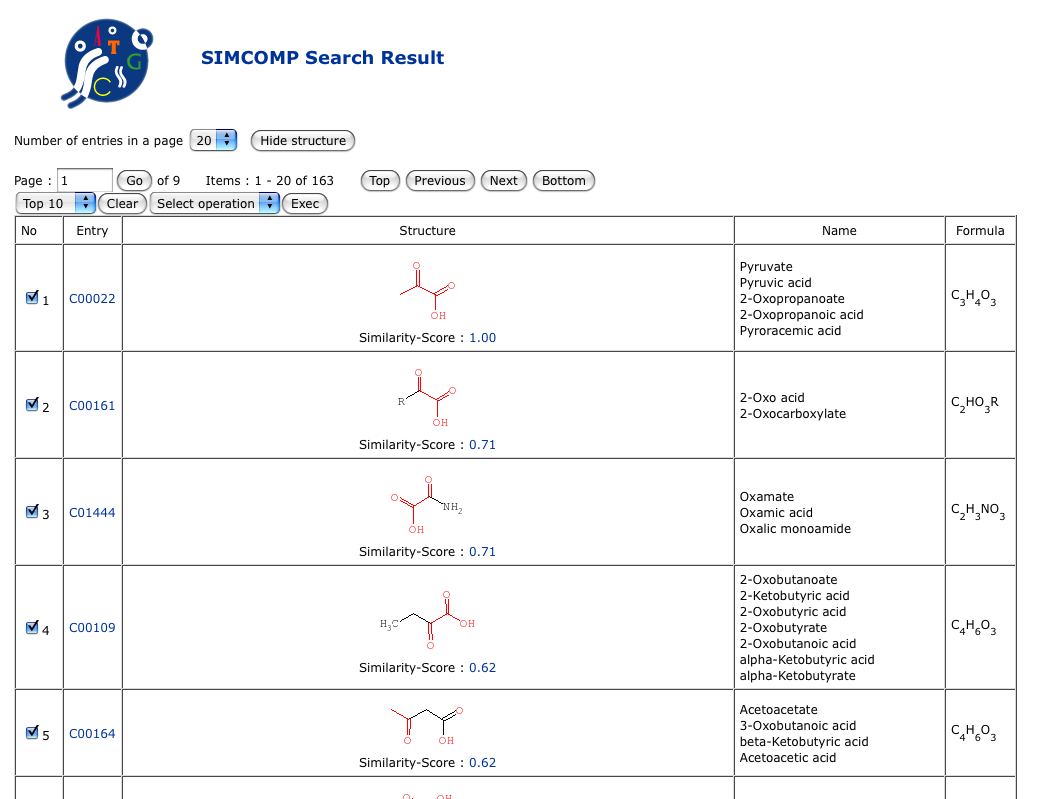
About SIMCOMPSIMCOMP (SIMilar COMPound) is a graph-based method for comparing chemical structures. It has been implemented in the KEGG system for searching similar chemical structures in the chemical structure databases.SIMCOMP uses an efficient algorithm for comparing two chemical compounds, where the chemical structure is treated as a 2D graph consisting of atoms as vertices and covalent bonds as edges. It is based on the algorithm to solve the maximal common subgraphs of two graphs as the maximum vertex induced common subgraph (MICS) or as the maximum edge induced common subgraph (MECS). Here, maximal common subgraphs of two graphs can be found by searching for maximal cliques in the association graph, and we have introduced heuristics to accelerate the clique finding. (see reference)
SIMCOMP provides the atom alignments between two chemical compound graphs, then it can also calculate the similarity of two chemical compounds by counting the number of matched atoms in those atom alignments.(see reference) For all calculations in SIMCOMP, the KEGG Atom Types are needed as a representation of atoms to detect biochemically meaningful features. The KEGG Atom Types are based on the concept of functional groups in chemistry, and 68 atom types (vertex types) have been defined for carbon, nitrogen, oxygen, and other atomic species with different environments. The current version of GenomeNet structural search provides the availabilities for MDL/Mol file format and SMILES format. When those formats are input, they will be transformed into KCF formats internally. KCF format and KEGG Atom TypesKCF (KEGG Chemical Function) format is a format of chemical objects like chemical compounds, glycan structures, and reactant pairs, which has been defined by KEGG. In particular case of chemical compounds, every atoms are represented as one of KEGG Atom Types in order to distinguish functional groups and microenvironments of atoms. The current KEGG Atom Types has 68 types which are compiled from organic atoms like C, N, O, S, and P and other atomic species. They were first introduced for detecting biochemical similarities by graph-based chemical structure comparison.How to use the query formHow to use the advanced optionsSIMCOMP offers advanced search options from "Option details", which will be shown when clicking the small triangle ( ). ).
"Docking mode" means which algorithm will be applied, "Atom based" (MICS based) approach or "Bond based" (MECS based) one. When selecting atom based or bond based algorithm, the docking graph will be made based on the condition of every atom-to-atom matches or bond-to-bond matches, respectively. Here a bond-to-bond match will be tested by checking all possibilities of matching four atoms connected by each bond. This means the bond based method is stricter than the atom based, resulting in the smaller docking graph and the faster clique finding. In usual cases, the computation will be about tenth faster than the atom based. However, such a result like matching of only one atom will never been obtained. The node condition of the docking graph can be controlled by selecting one of three: (1) "Atom species" for testing by the element like 'C', (2) "Atom class" for the classification of KEGG Atom Types, like 'C1', or (3) "KEGG atom" for using the whole notation of KEGG Atom Types, like 'C1a'. Note "KEGG atom" is the most strict and results in the smallest docking graph and the fastest computation. "Post-processing mode" means whether the post-processing treatment will be applied or not after the clique finding. When selecting "None", no post-processing will be applied. Otherwise, the post-processing will be done by eliminating and/or extending simply connected common subgraphs (SCCSs). Here "for the largest SCCS" is for eliminating other SCCSs except the largest one and extending it. "for all SCCSs" is for extending all SCCSs without any elimination. The node condition when extending SCCSs can be controlled in the similar way with above. Here this is recommended to be identical to or looser than the above node condition, because the region with stricter node condition might be found at the clique finding step beforehand and it is expected to have already been included in SCCSs. SIMCOMP will detect the chirality change between query compound and database entry, provided that the information of chirality is properly given. Here the R-/S- chirality of asymmetric carbons can be designated as the up- or down- arrows on the 2D graph, and the cis-/trans- chirality around C=C double bond should be described with proper coordinates on 2D plane. When the chirality change is detected, the resulting similarity scores will be deducted the amount of 0.1 atom match per change to distinguish the isomers. If the "Chiral check" option is set to off, these chiral detection features will be turned off. Further operationAfter obtaining the search result, the further computation features are available by selecting an item from "Select operation" menu and clicking the "Exec" button. Here, to choose "Map to Pathway" or "Map to BRITE" mean to search checked entries for PATHWAY database or BRITE database, respectively.About SIMCOMP2SIMCOMP2 provides all vs. all computation of chemical structure similarity between two sets of user specified compounds. The input sets can be either KEGG compound/drug IDs or MOL file texts. When you use KEGG IDs, it can be a search result including definitions and other comments that will be automatically removed from the input. You can see how it works by copying & pasting the search result such as this and clicking button.The search results are limited to those with similarity scores 0.8 or higher by default. You can change this cut off score in the Options details section, as well as the other options for the SIMCOMP search. References - Hattori, M., Tanaka, N., Kanehisa, M., and Goto, S.; SIMCOMP/SUBCOMP: chemical structure search servers for network analyses. Nucleic Acids Res. 38, W652-W656 (2010). [pubmed] - Hattori, M., Okuno, Y., Goto, S., and Kanehisa, M.; Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. J. Am. Chem. Soc. 125, 11853-11865 (2003). [pubmed] - Hattori, M., Okuno, Y., Goto, S., and Kanehisa, M.; Heuristics for chemical compound matching. Genome Informatics 14, 144-153 (2003). [pubmed] Last updated: August 20, 2010 « Back |