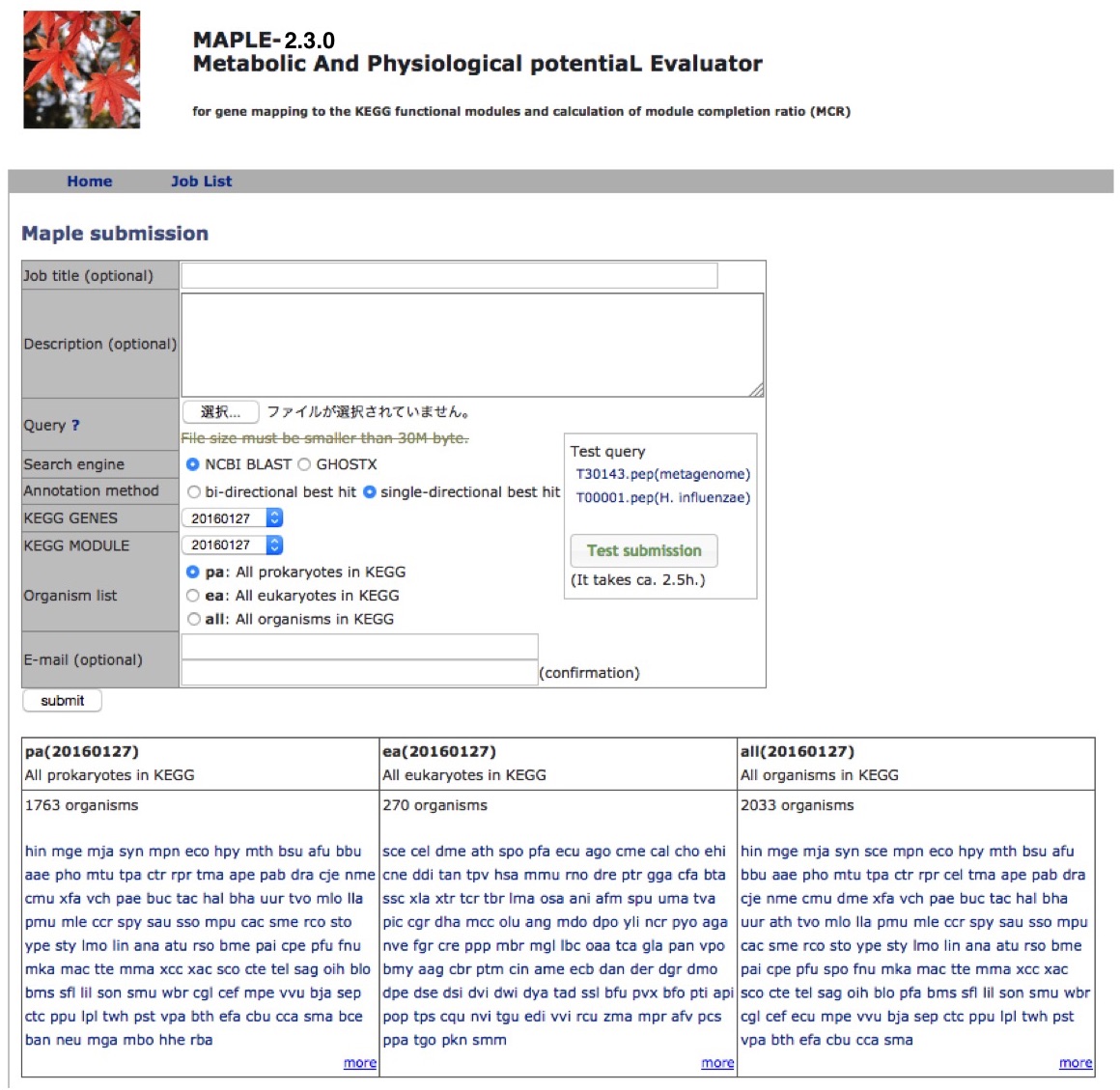
MAPLE HelpMAPLE (Metabolic And Physiological potentiaL Evaluator) is an automatic system for mapping genes in an individual genome and metagenome to a functional module and for calculating the module completion ratio (MCR) in each functional module defined by Kyoto Encyclopedia of Genes and Genomes (KEGG). First, this system assigns KEGG orthology (KO) to the query genes using the KEGG automatic-annotation server (KAAS) and maps the KO-assigned genes to the KEGG functional modules automatically. After mapping genes to modules, MAPLE automatically calculates MCR of each functional module. MAPLE can display results of comparative analyses of mapping patterns, MCR results, and abundance of completed modules not only among KEGG-annotated genomes but also among user jobs as well as between user jobs and KEGG-annotated genomes. Data Submission Query sequences
A metagenome
An example of a dataset: complete or incomplete genes predicted from short read sequences
Organism's data selection
Database selection
Homology search tool selection
An individual organism
An example of a data set: complete genes Notification of MAPLE job completion
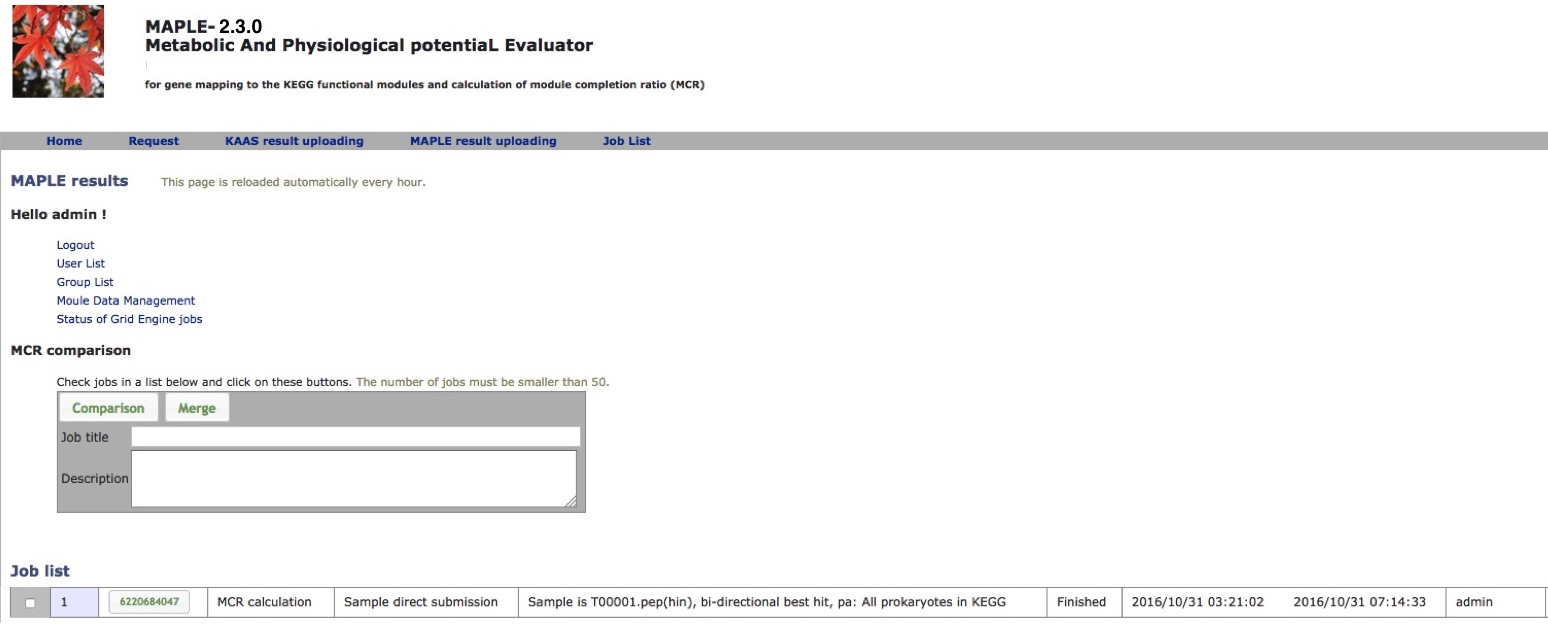
After submission of the dataset, the following message will be displayed (Figure 2): "The URL of results will be sent to the registered user's e-mail address after all processes are completed" and a job list will be present on the result page (Figure 3).
MAPLE Result Page
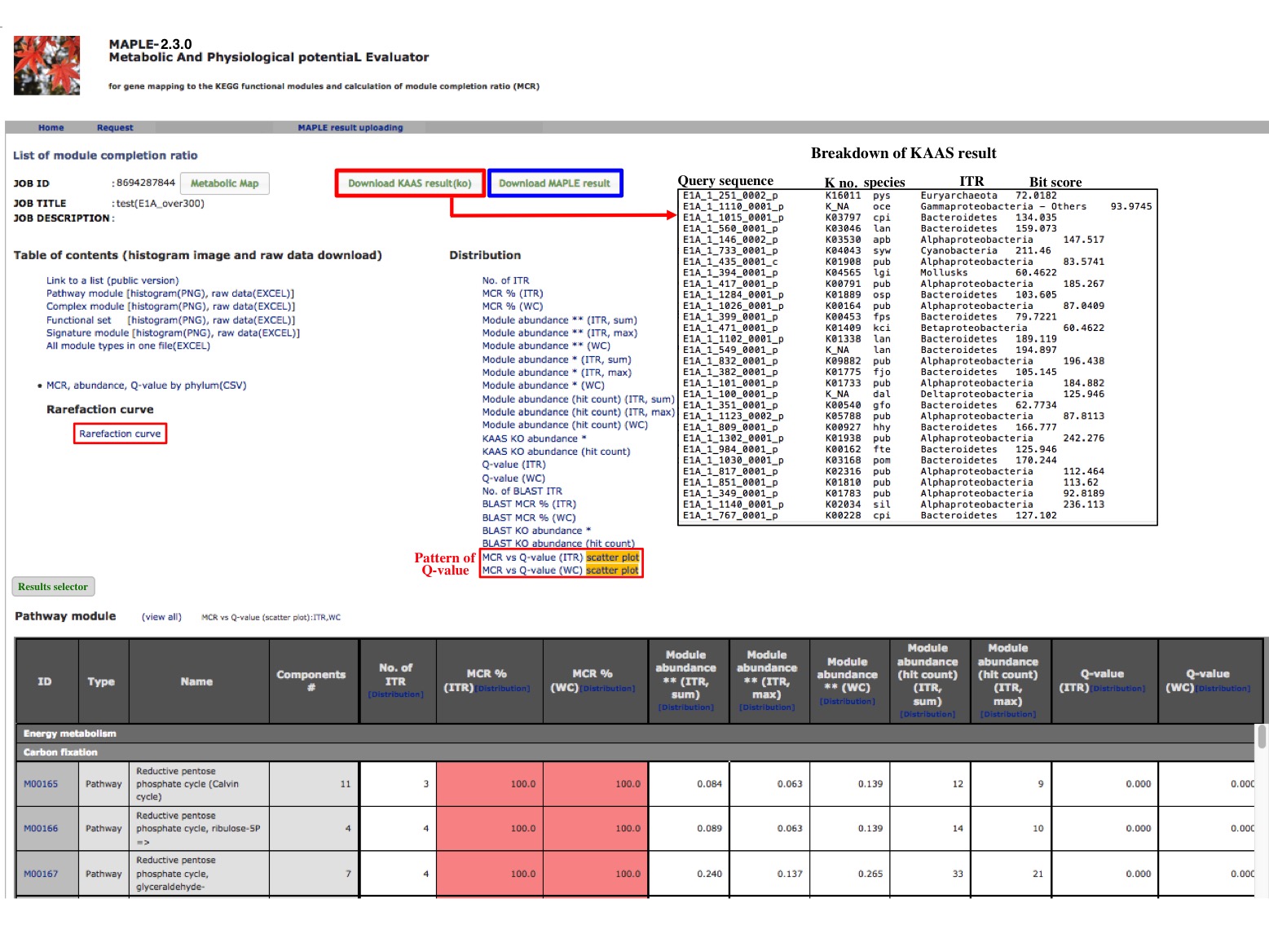
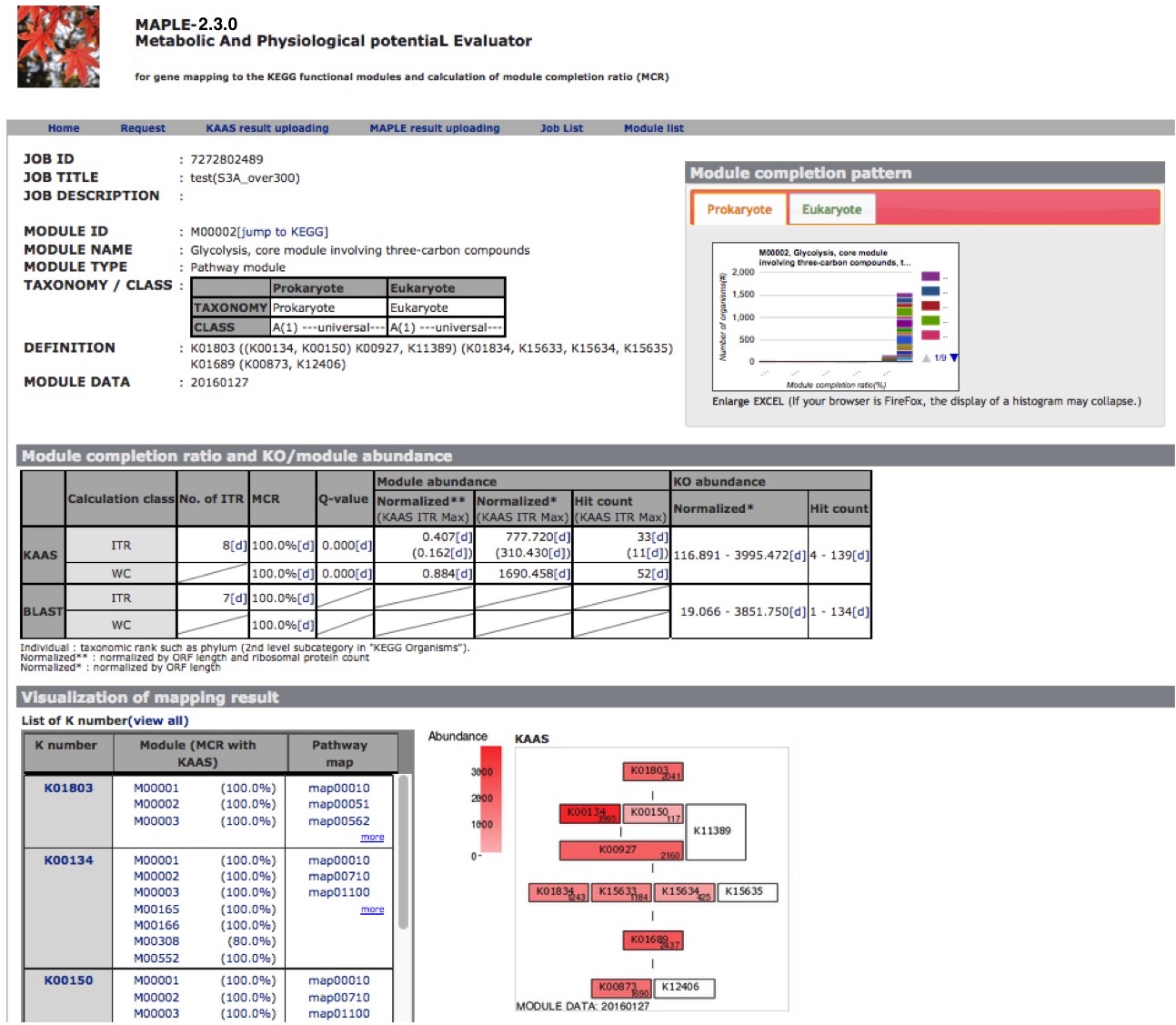
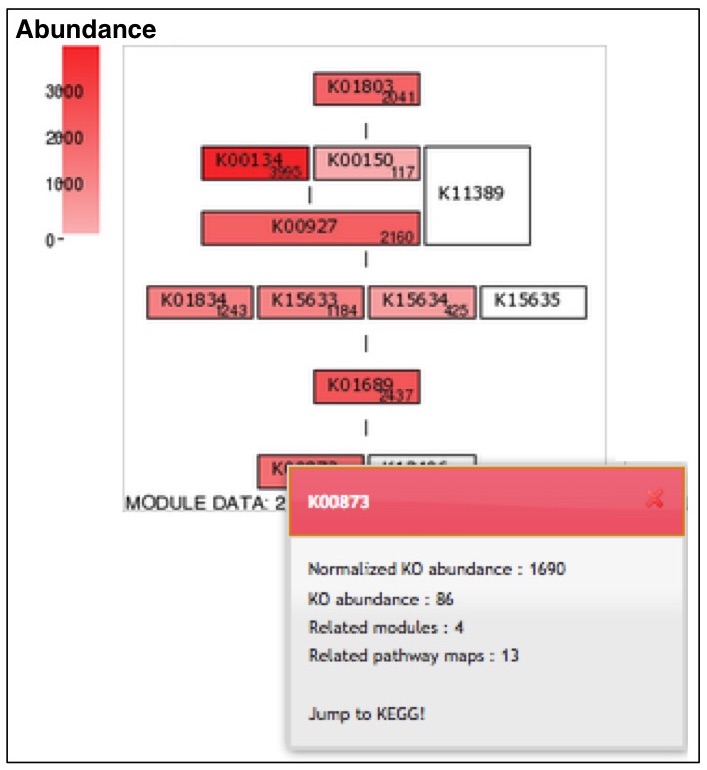
After completion of the calculation, the job ID is available for further interpretation of results (Figure 3). Clicking the job ID yields MCR (ITR for individual taxonomic rank and WC for whole community), module abundance (ITR and WC), and Q-value (ITR and WC) for determining the significance of module completeness (Figure 4). MAPLE system can yield various results by every ITR such as Gammaproteacteria, Actinobacteria, and Cyanobacteria and also by WC (whole community) (see ref. 1 for details). Users can select individual results, which appear on the result page depending on necessity from the display selector of the results (Figure 4). Users can download these data in Excel format and view all MCRs for the KEGG modules as a histogram on the first page. In addition, users can download the list of KO-assigned sequences generated by KAAS by clicking "Download KAAS results (KO)" (red rectangle). Users are advised to download the job file to reproduce all MAPLE results because each user's job will be automatically removed from the job list two weeks later. If the user clicks "Download MAPLE results" (blue rectangle), a compressed file can be downloaded. If the user uploads this file later, all MAPLE results will be reproduced on the web page. If the user clicks a module ID, the mapping pattern of the KEGG module is displayed as shown below.
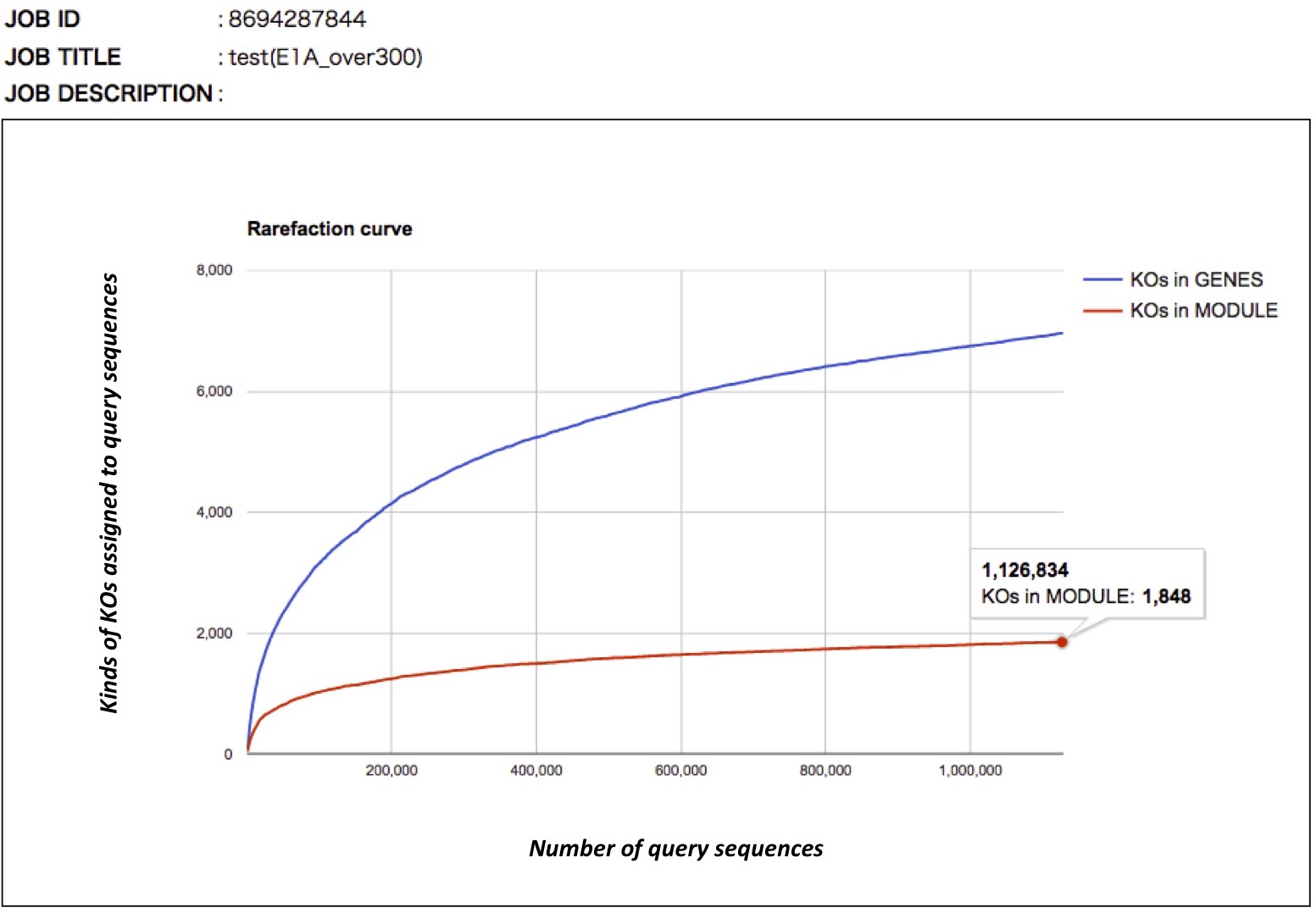
Rarefaction curve
Q-value for determining the significance of module completeness
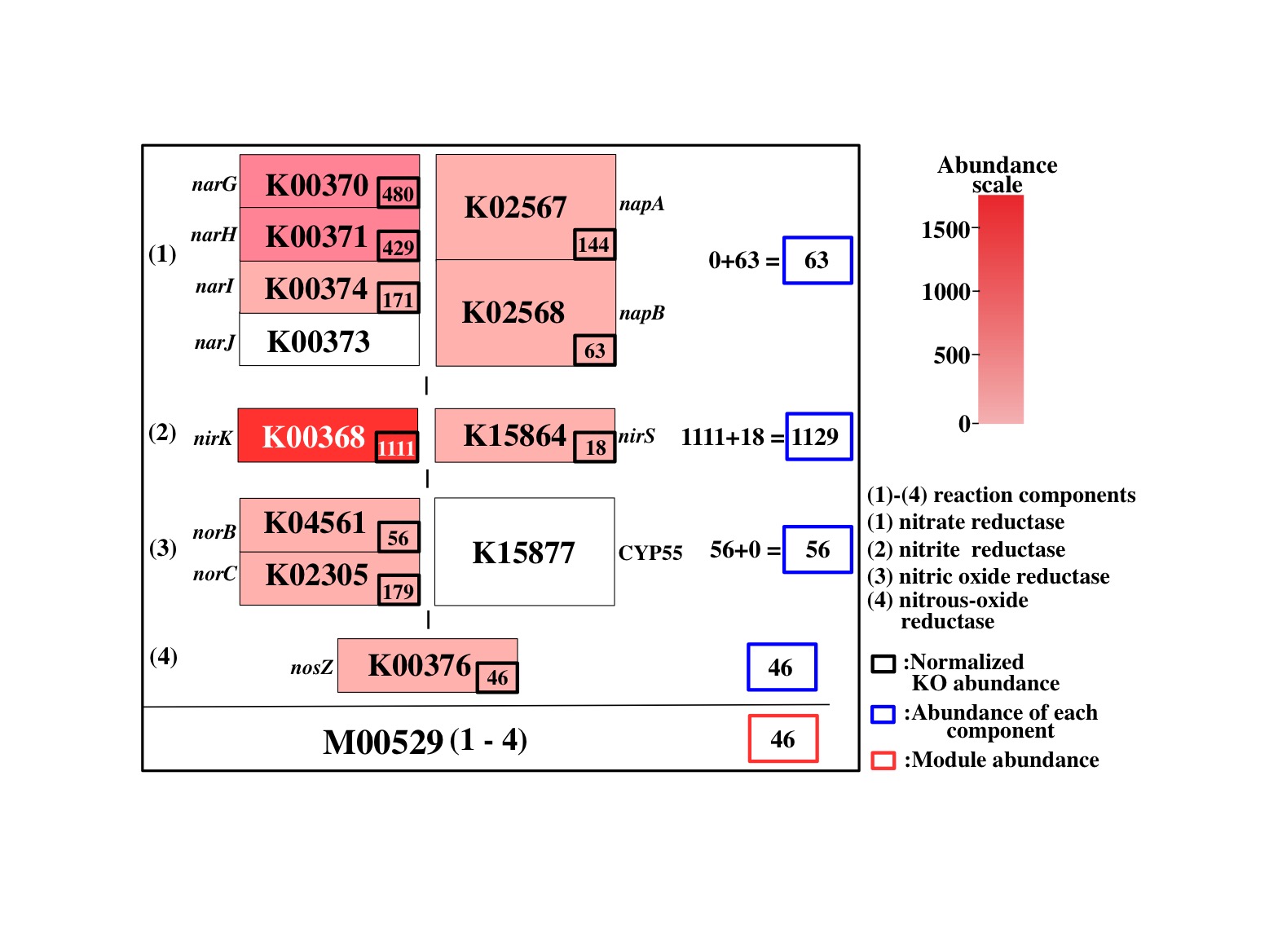
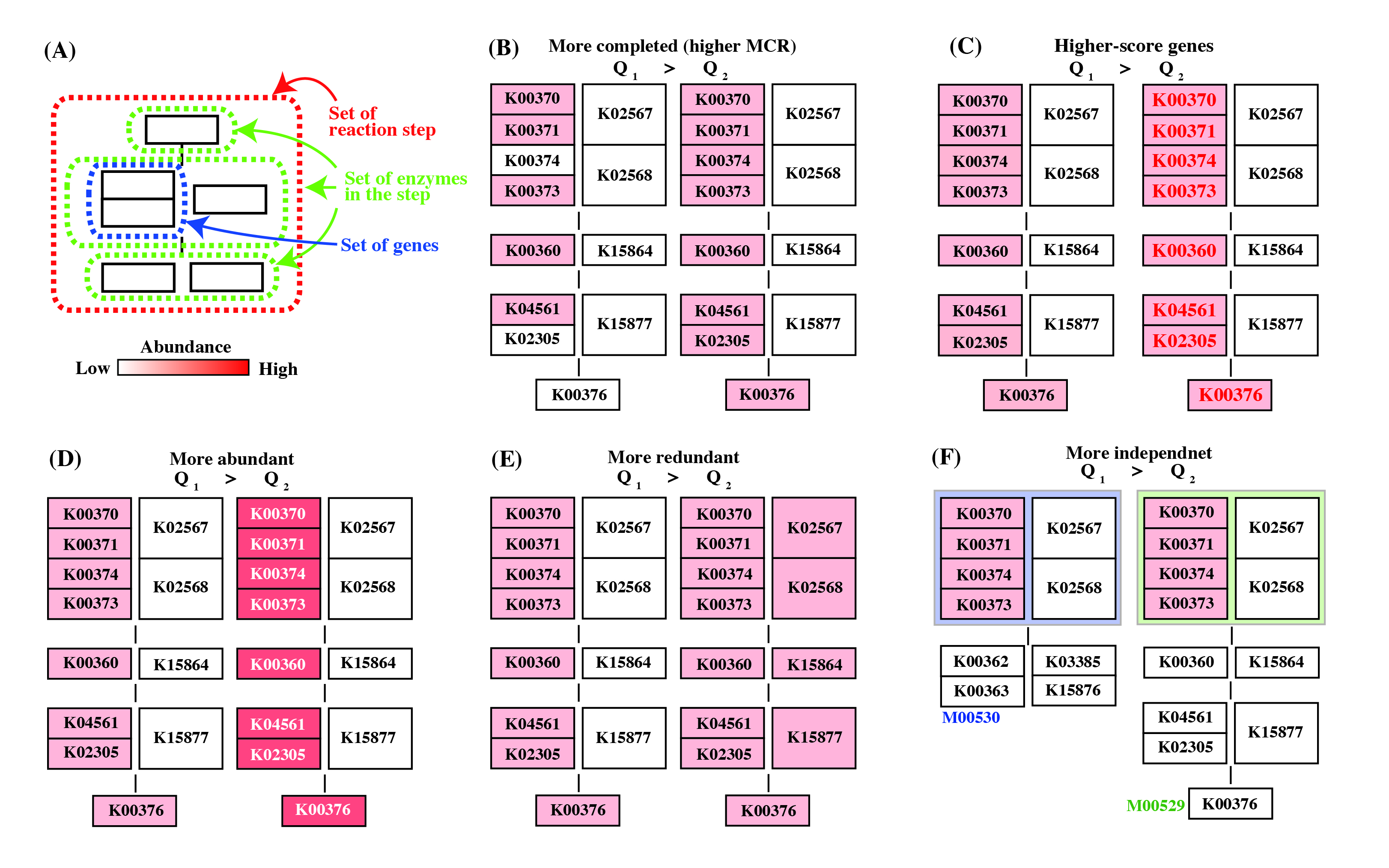 Figure 6. General concept of Q-value. Click to enlarge. (A) Schematic diagram of reaction module. (B) - (E) As an example, the reaction module M00529 is shown. The weight of the K number (e.g., K00370) indicates the sequence similarity scores. The Q-value is lower in the following cases. (B) The module is more completed (i.e., it has a higher MCR). (C) The module consists of genes with higher similarity scores. Red numbers mean high similarity score. (D) Genes are more abundant. (E) Enzymes or reactions are alternative (e.g., isozymes exist). (F) The module is less overlapped with the other modules because of a fewer multiple comparisons. Note that the left-side module in (F) is ideal (i.e., M00529 is assumed to be independent from M00530) for the purpose of illustration.
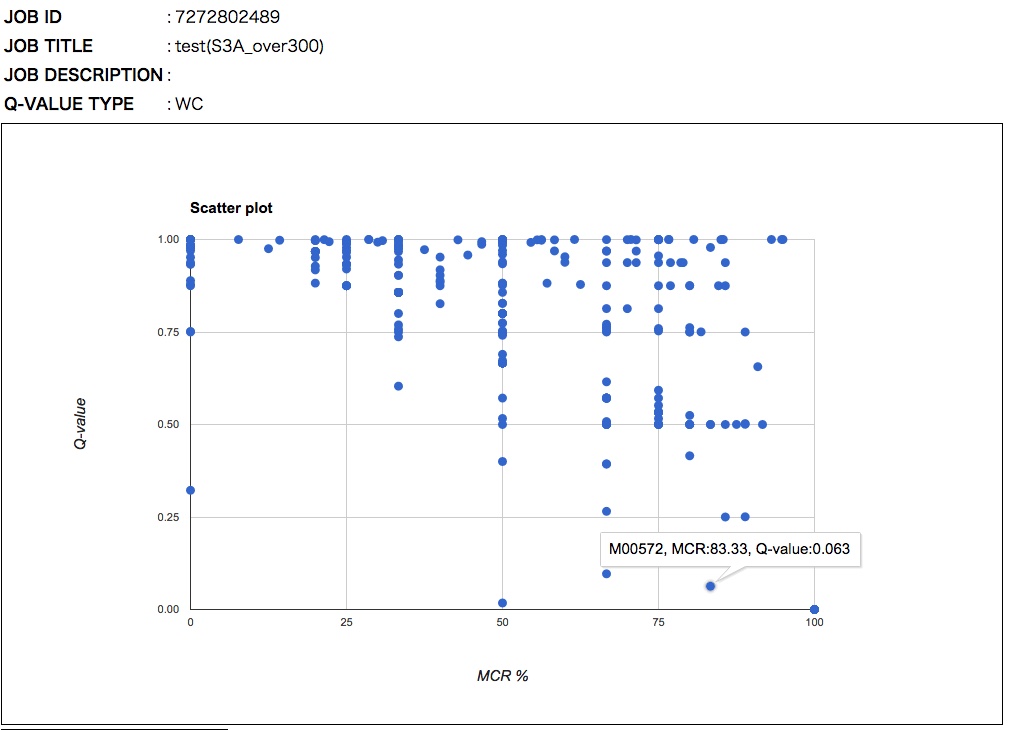
The general features of the Q-value are summarized in Figure 6. The Q-value decreases with increasing MCR and becomes smaller when genes are more abundant and when alternate reactions are available. Reaction modules that are more frequently overlapped with other modules and longer reaction modules exhibit higher Q-values because of multiple testing corrections. According to definition of the Q-value, its value is larger than 0.5 when a straightforward (simplest or most fragile) reaction module has at least one unidentified KO (i.e., MCR < 100 %). Thus, as a criterion, we can interpret that reaction modules with Q-values of less than 0.5 are biologically feasible when the MCR is less than 100%. The patterns of Q-value to MCRs of all modules (WC and ITR) are shown in each job (Figure 7) by clicking "MCR vs Q-value" on the result page (Figure 4). Module Completion Ratio (MCR) pattern
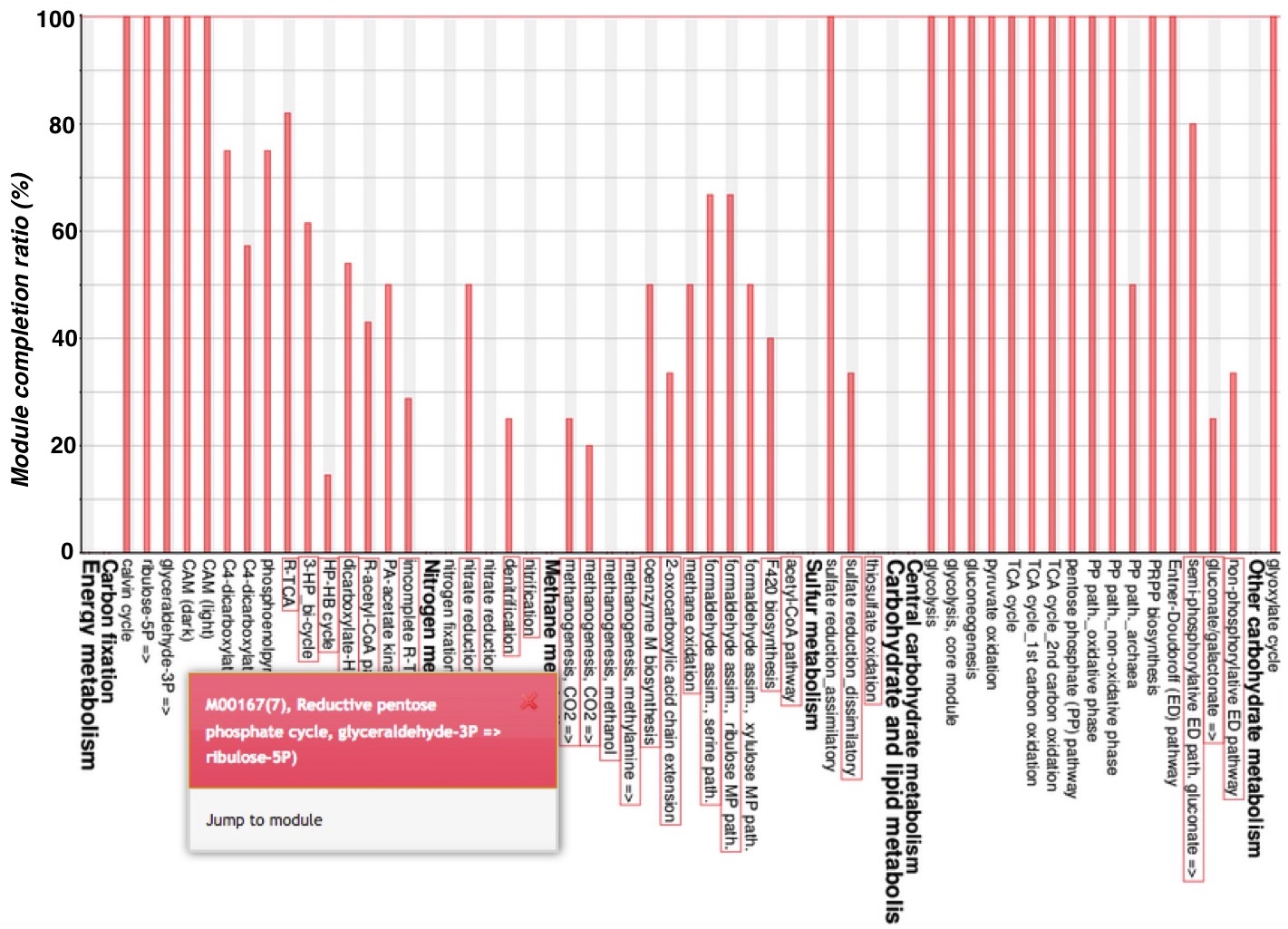
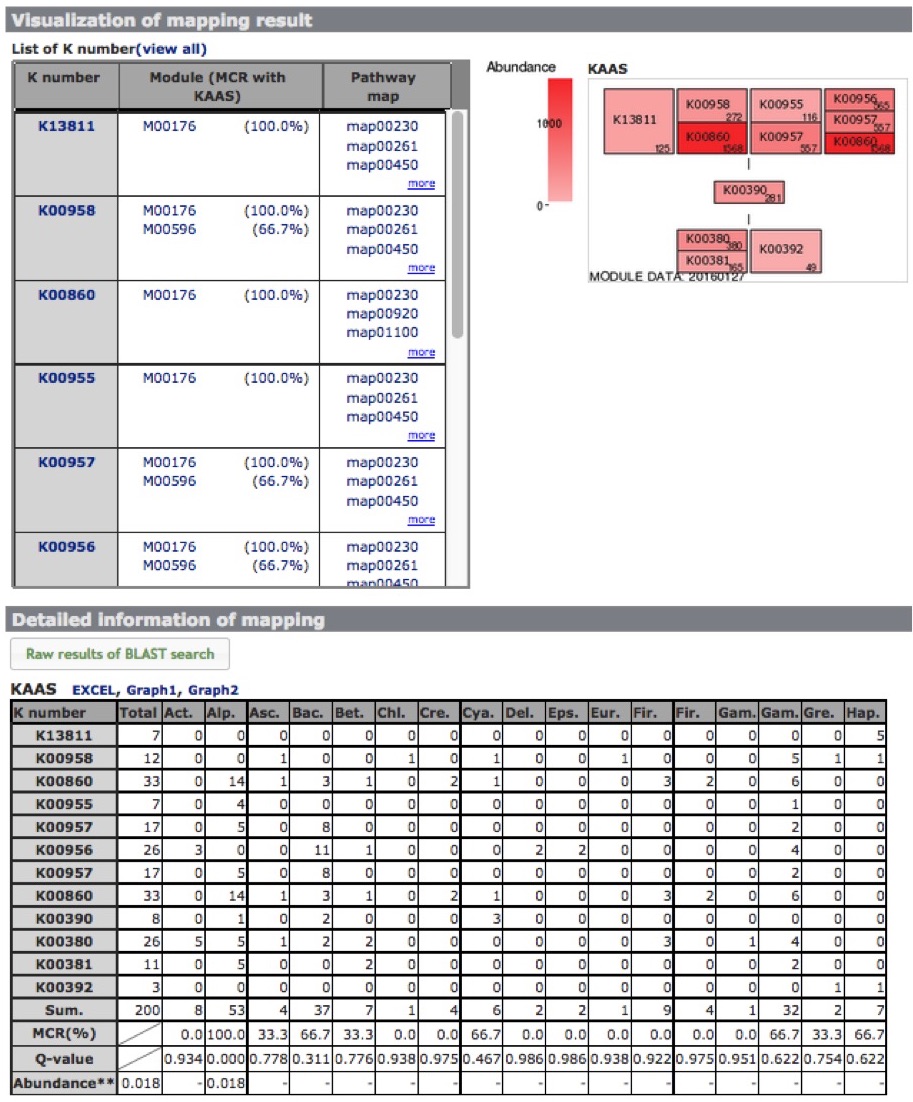
The module completion pattern of reference organisms Distribution of MCRs in 1488 prokaryotic or 259 eukaryotic species (one genome per species) can be categorized into four patterns (universal, restricted, diversified, and nonprokaryotic/ noneukaryotic) regardless of the module type (pathway, structural complex, signature, or functional set), considering 70% of all species represent a majority measurement for patterns. Users can easily select a prokaryotic or eukaryotic pattern (Figure 9). Pattern A, defined as "universal" consists of modules completed by more than 70% of all prokaryotic or eukaryotic species. Pattern B, defined as "restricted" consists of modules completed by less than 30 % of species, with rare modules completed by less than 10% of all species. Pattern C, defined as "diversified" consists of modules ranging widely in MCR. Pattern D, defined as "nonprokaryotic" or "noneukaryotic" consists of modules that are not completed by prokaryotic or eukaryotic species (Figure 10). These four criteria and taxonomic classification for each module are helpful for interpretation of module completion patterns. 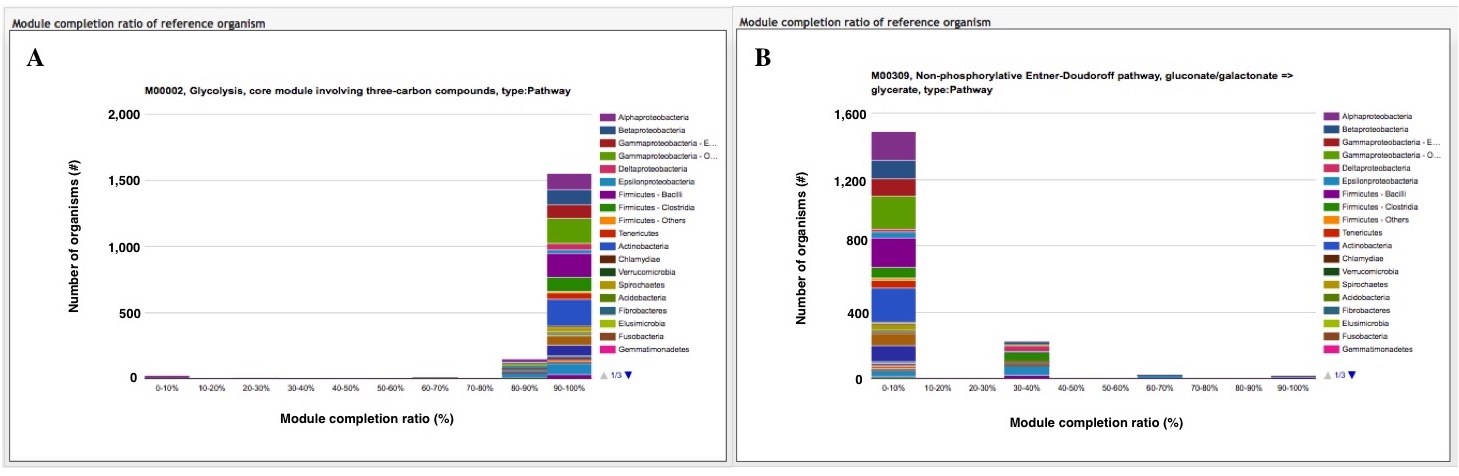 Figure 10. Module completion pattern for the KEGG modules from 1763 prokaryotic species. Click to enlarge. (A) The module that is completed for almost all reference organisms. (B) The module that is completed for a limited number of organisms. The breakdown of organisms for which the module is completed is color-coded by phylum level taxonomy. The list of K numbers Some KO identifiers (K numbers) that are assigned to a KEGG module are shared by several other modules. These modules are listed with MCR (Figure 11). When MCR is low, the relationship between MCR of the targeted module and others assigned the same K number should be considered. In addition, all pathway maps possessing the K number assigned to the module are presented in the rightmost column. A taxonomic breakdown of sequences assigned K numbers is shown below the list of K numbers. The list from the taxonomic breakdown can be downloaded in Excel format by clicking the button "Raw results of BLAST search" (Figure 11). The mapping pattern
The module abundance
When we compare the module abundances with those of other metagenomic samples, each sample should be normalized because the number of cells used to construct the metagenome is different depending on the sample, even if the number of sequence reads applied to MAPLE is the same. The ribosome is basically composed of the same number of ribosomal proteins, regardless of the species, although some species possesses accessory proteins and almost all genes encoding ribosomal proteins are single copy, i.e., the number of ribosomal proteins in the metagenome reflects number of cells. In MAPLE system, we calculated the module abundance per ribosomal protein to facilitate the comparison between the different environmental sites (see Ref. 1 for details).
Virtual module for ribosome  Figure 14. Virtual ribosome modules for taxonomic analysis in the metagenome. Click to enlarge. A: M90000 (virtual prokaryotic module), B: M91000 (virtual module for all organisms)
Comparison of multiple results
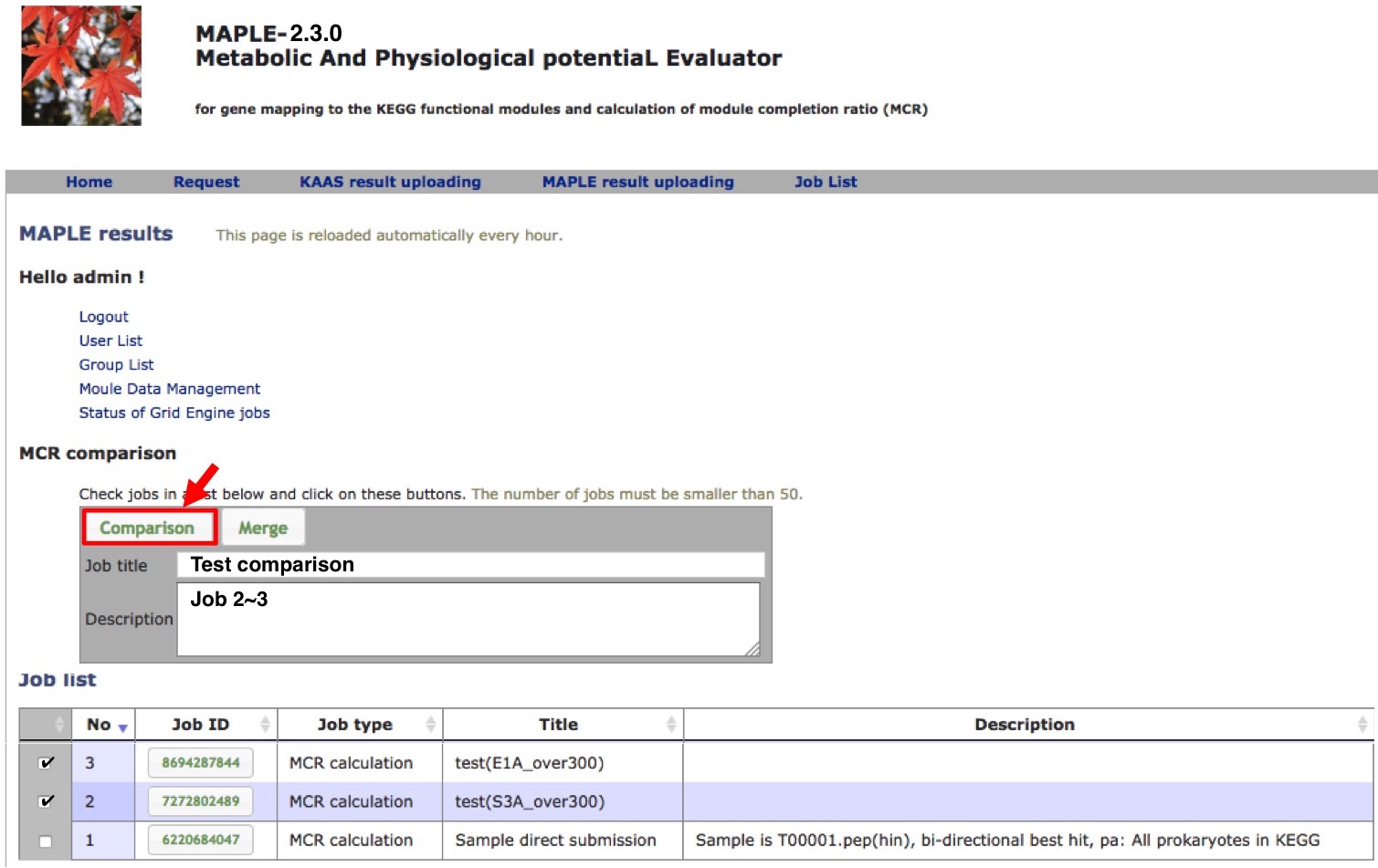
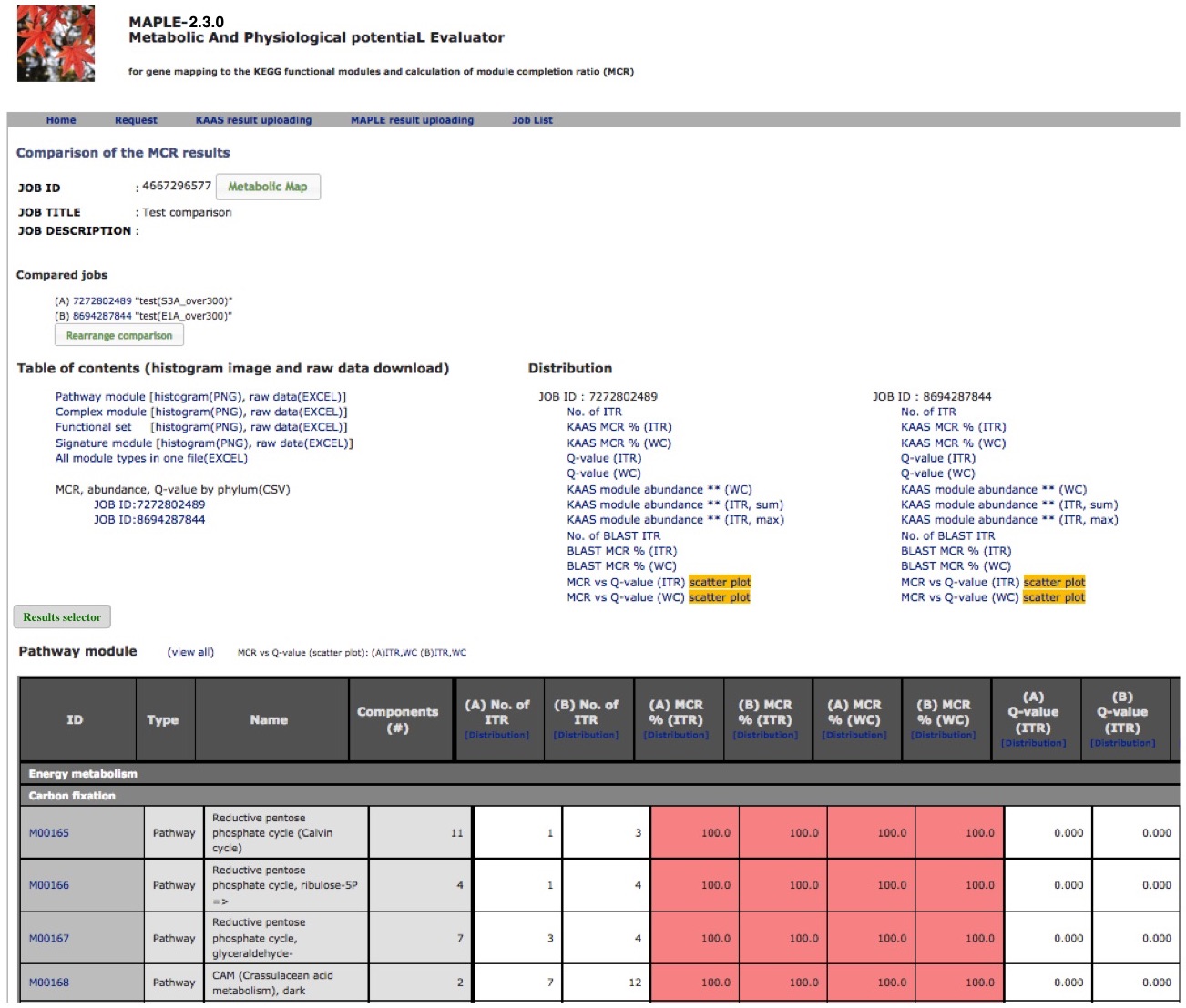
Comparison of several custom jobs 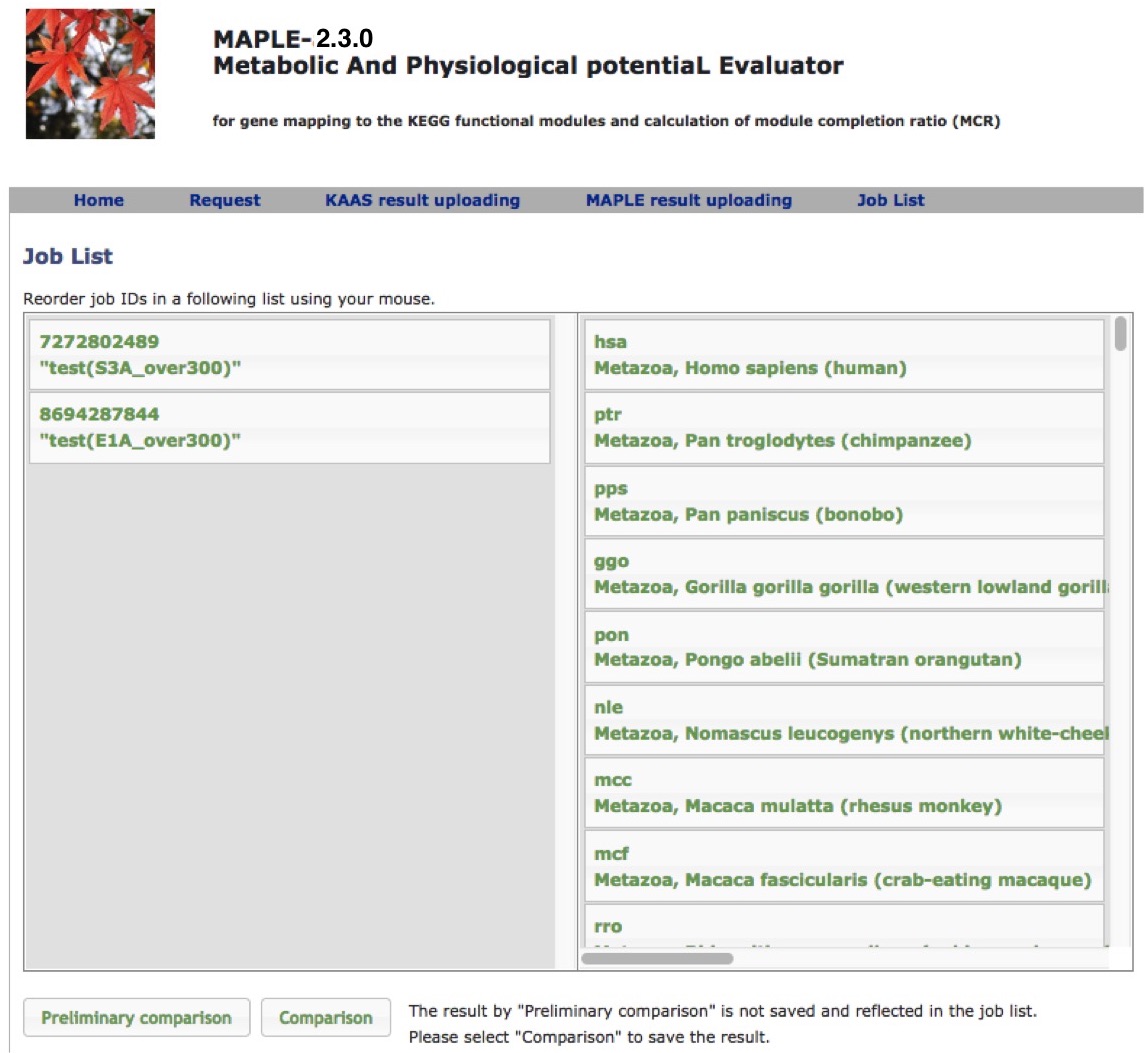 Figure 16. Job arrangement page for comparative analysis Click to enlarge. 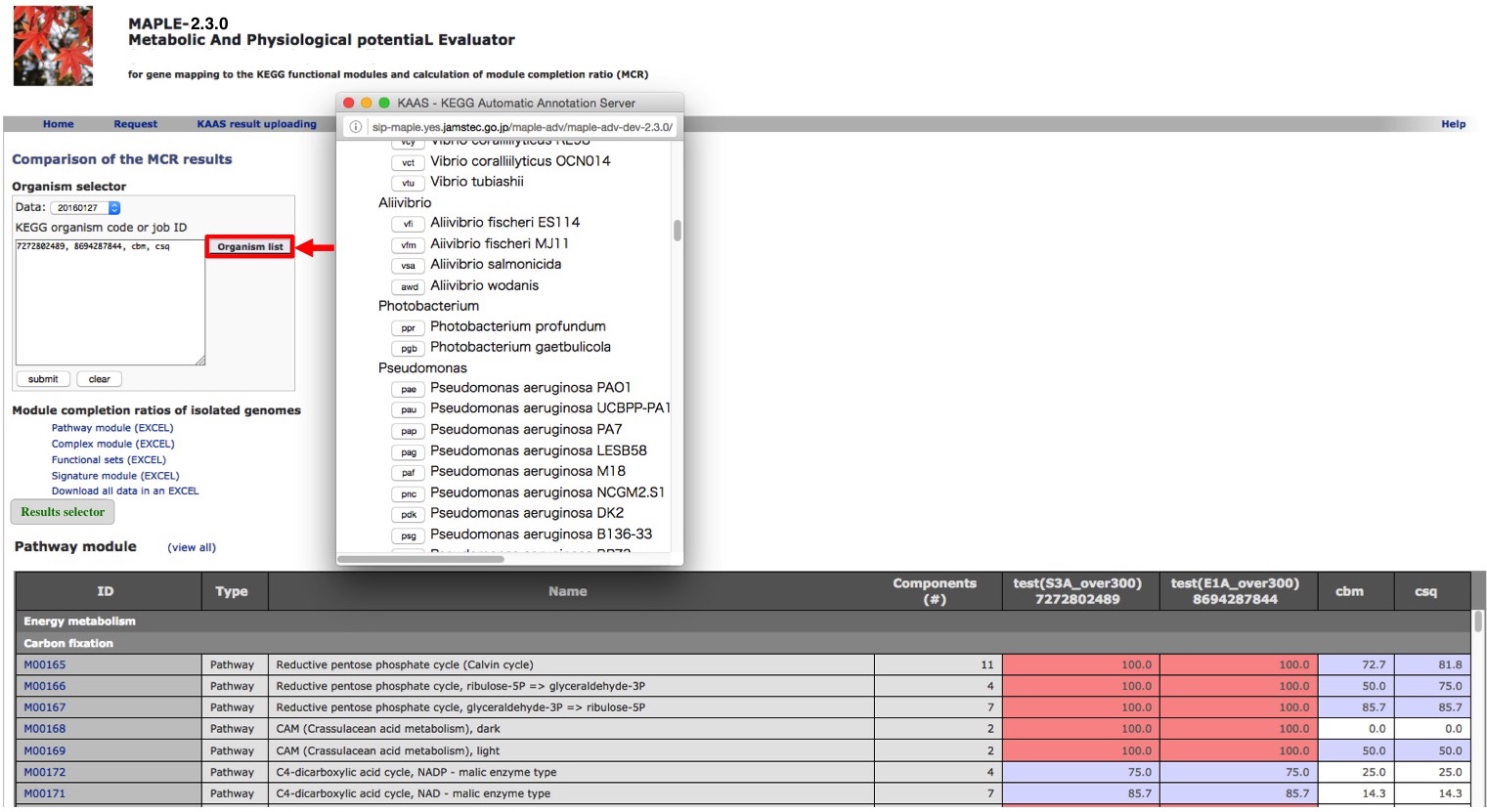 Figure 17. Results of the preliminary comparison Click to enlarge.
Preliminary comparison
Comparison and saving results
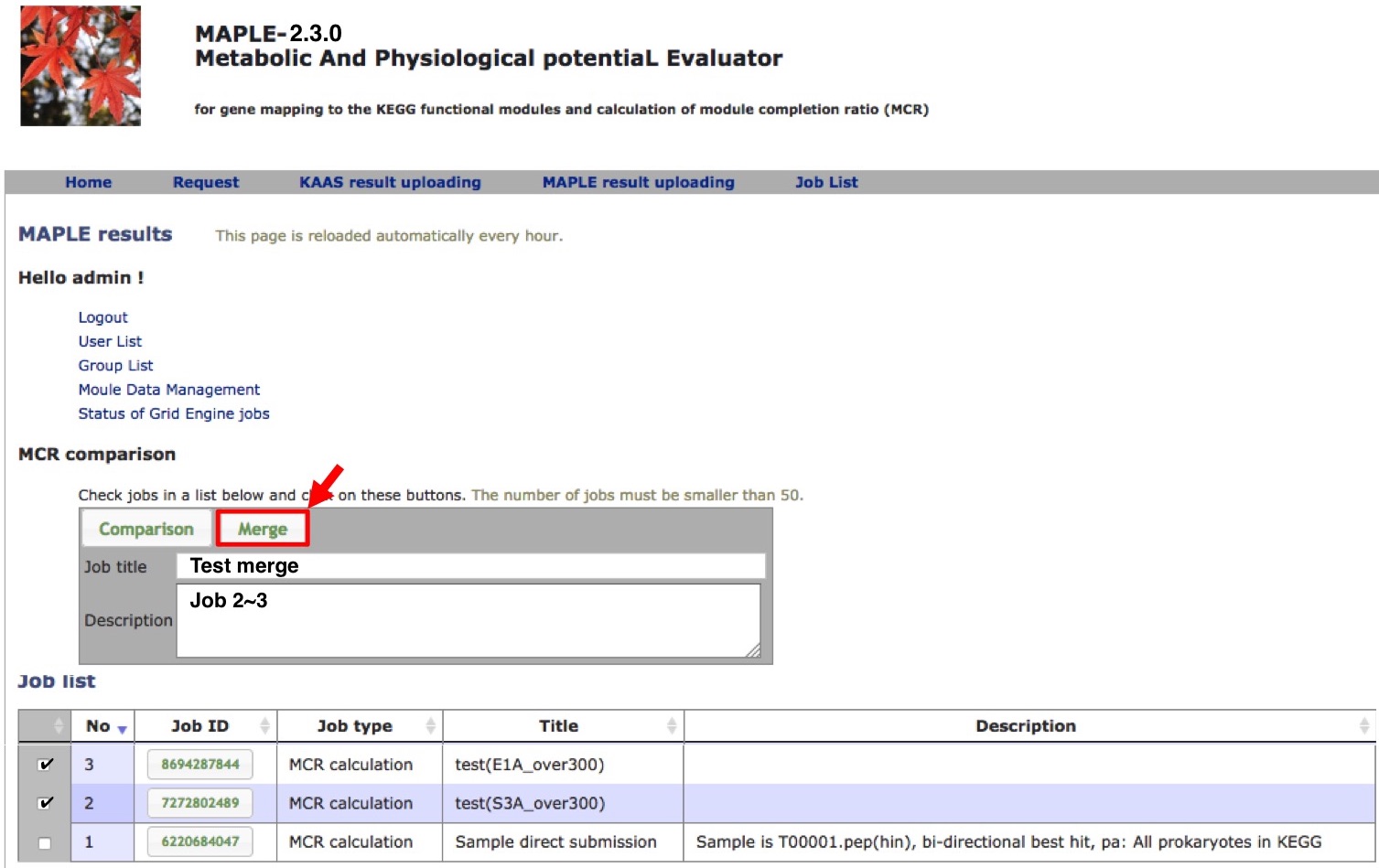
Merging results of subdatasets
Uploading MAPLE results |