|
The latest version of MAPLE is available at JAMSTEC. About MAPLEMAPLE (Metabolic And Physiological potentiaL Evaluator) is an automatic system for mapping genes in an individual genome and metagenome to the functional module and for calculating the module completion ratio (MCR) in each functional module defined by Kyoto Encyclopedia of Genes and Genomes (KEGG). The MCR calculation is performed based on a Boolean algebra-like equation defined by KEGG to each module. MAPLE first assigns a KO identifier (ID) to the query gene using KAAS, maps the KO-assigned genes to the KEGG functional modules, and calculates the MCR of each functional module and its abundance when the module is complete. There are two methods for KO assignment by KAAS: bidirectional best hit (BBH) and single-directional best hit (SBH). The BBH method is suitable for complete gene sets identified in complete genomes or contigs, while the SBH method is mainly for short-read sequences in metagenomes or incomplete genomes. The result page displays the MCR, abundance of each KEGG module and the taxonomic information of the KO-assigned genes mapped to the module along with a mapping pattern. Also, a module list sharing the same KOs is shown. The results of KO assignment by KAAS, taxonomic information of the genes mapped to the KEGG modules, and calculated MCRs are downloadable in an Excel format. MAPLE can display the results of comparative analyses of mapping patterns, MCR results, and abundance of complete modules between different metagenomic samples. Generally, it is expected that the MCR is linked to the likelihood that the organisms perform the physiological function corresponding to the module. However, when the KOs used for a module are shared with the other modules, the MCR does not necessarily reflect the working probability of each functional module. To evaluate the working probability of the physiological function in the incomplete modules, we proposed the Q-value for determining the significance of module completeness. The Q-value, which implies the probability that a reaction module is identified by chance, is calculated based on the statistics of the sequence similarity score and KO abundance using the concept of multiple testing corrections according to the Boolean algebra-like equations. Metagenome or Partial Genome SequencesKO assignment to short-read sequences (400–500 nt) produced by a high-throughput DNA sequencer is performed by KAAS using the single-directional best-hit (SBH) method. Query sequences must be translated into amino acid (aa) sequences before submission, and sequences longer than 100 aa are recommended for accurate KO assignment. Complete or Draft Genome SequencesKO assignment for complete gene sets that were identified in the complete genome or contigs is performed by KAAS using the bidirectional best-hit (BBH) method. Comparison of the KEGG‐Annotated GenomesComparison of KEGG-annotated genomes without user's jobs is also possible and does not require an e-mail address. KEGG Data used in MAPLEMAPLE uses the following KEGG GENES and MODULE databases:
History
|
Example of Results
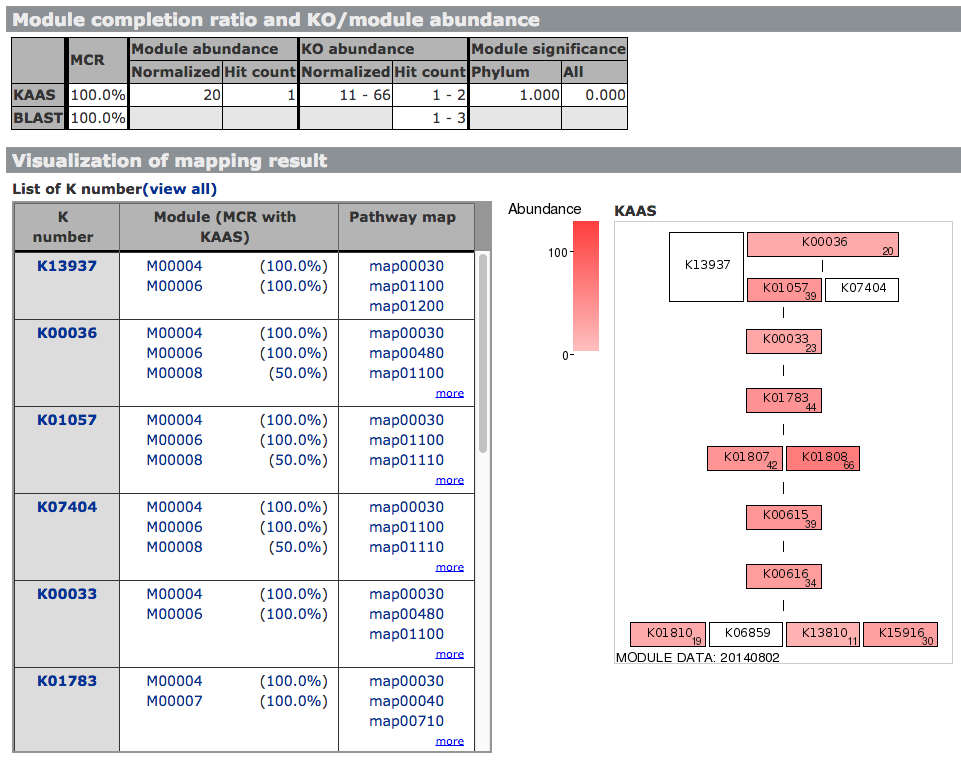
Mapping genes to the KEGG functional modules
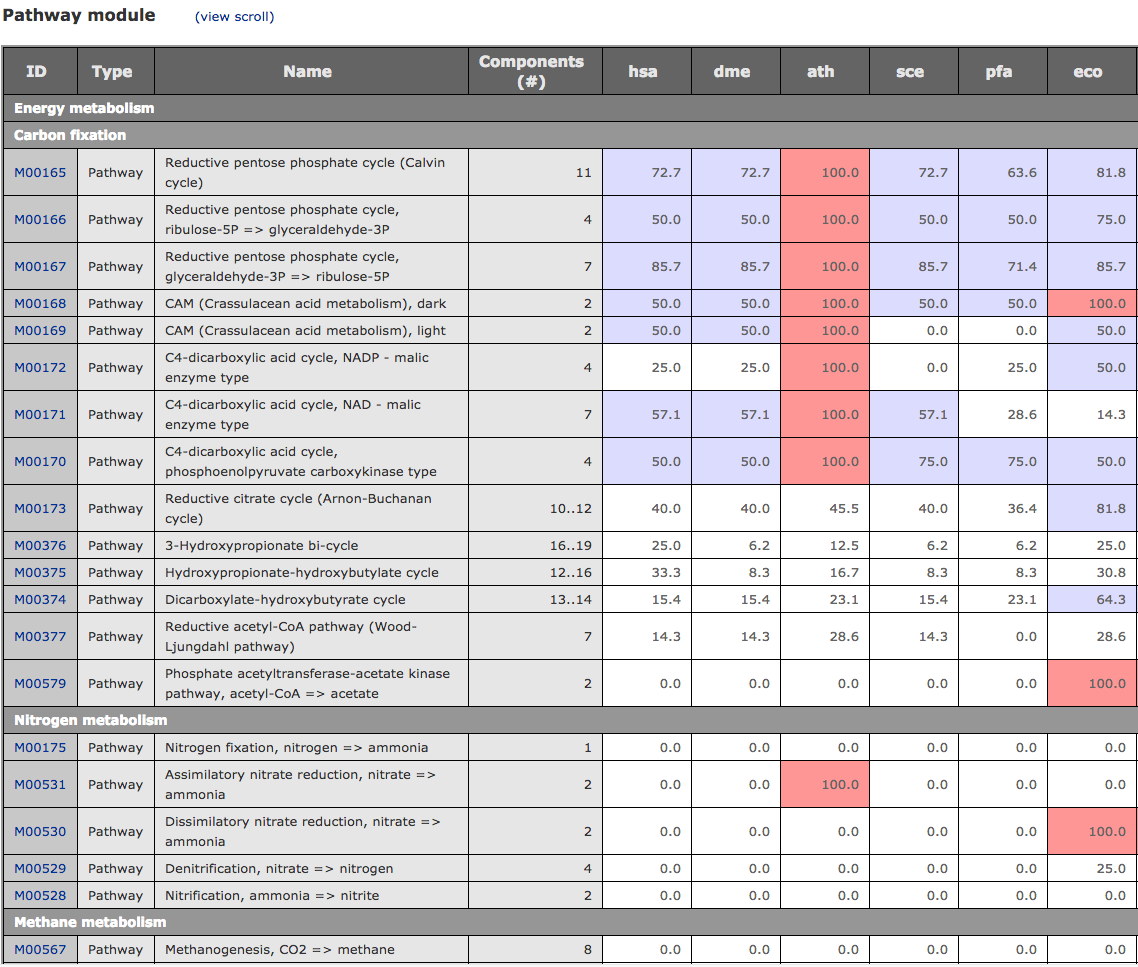
MCR calculation of each KEGG module
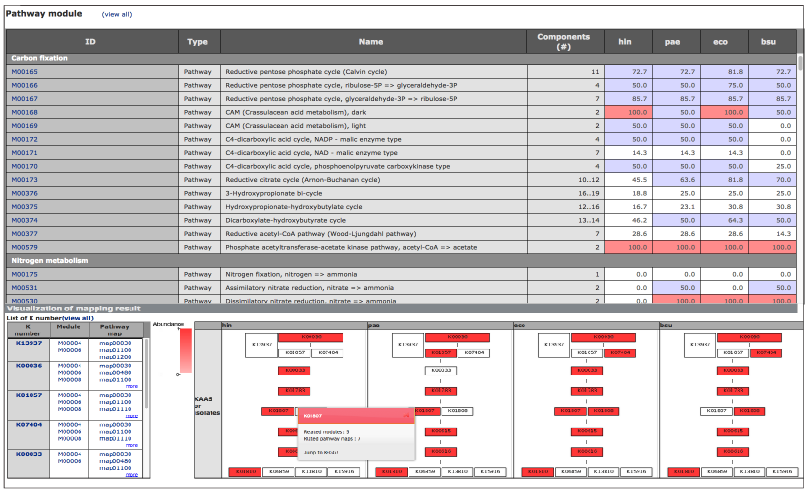
Results comparison
|
||||||||||||||||||
References
Last updated: Last updated: Nov. 25, 2016 |