
KAAS (KEGG Automatic Annotation Server) provides functional annotations of genes in a genome by amino acid sequence comparisons against a manually curated set of ortholog groups in KEGG GENES. The result contains KO (KEGG Orthology) assignments and automatically generated KEGG pathways and functional classifications. This server is used internally to annotate query genes for the MAPLE functional evaluator.
Query form
Search program
The following four homology search programs are available for the initial screening to obtain similar sequences from the target database.
- BLAST - GHOSTX - GHOSTZ
In general, GHOSTX is much faster than BLAST using a suffix array indexing, but is less accurate. GHOSTZ uses a compressed database and is further faster, but is much less accurate. We recommend to use GHOSTX when you have a large number of query sequences or use a large target data set.
Query sequences
Normally query sequences are amino acid sequences representing a set of protein-coding genes in a complete genome. KO assignments are based on results from the homology search tools listed above, where BLAST also allow nucleotide sequences as queries.
Check the "Nucleotide" checkbox if queries are nucleotide sequences representing a set of EST contigs or ESTs. In this case, KO assignments are based on results from BLASTX and TBLASTN.
In both cases sequences should be in multi-FASTA format with unique IDs that should not include tab characters.
ex.)
>0001
MVKVYAPASSANMSVGFDVLGAAVTPVDGALLGDVVTVEAAETFSLNNLGRFADKLPSEP
RENIVYQCWERFCQELGKQIPVAMTLEKNMPIGSGLGSSACSVVAALMAMNEHCGKPLND
TRLLALMGELEGRISGSIHYDNVAPCFLGGMQLMIEENDIISQQVPGFDEWLWVLAYPGI
KVSTAEARAILPAQYRRQDCIAHGRHLAGFIHACYSRQPELAAKLMKDVIAEPYRERLLP
GFRQARQAVAEIGAVASGISGSGPTLFALCDKPETAQRVADWLGKNYLQNQEGFVHICRL
DTAGARVLEN
>0002
MKLYNLKDHNEQVSFAQAVTQGLGKNQGLFFPHDLPEFSLTEIDEMLKLDFVTRSAKILS
AFIGDEIPQEILEERVRAAFAFPAPVANVESDVGCLELFHGPTLAFKDFGGRFMAQMLTH
IAGDKPVTILTATSGDTGAAVAHAFYGLPNVKVVILYPRGKISPLQEKLFCTLGGNIETV
AIDGDFDACQALVKQAFDDEELKVALGLNSANSINISRLLAQICYYFEAVAQLPQETRNQ
LVVSVPSGNFGDLTAGLLAKSLGLPVKRFIAATNVNDTVPRFLHDGQWSPKATQATLSNA
MDVSQPNNWPRVEELFRRKIWQLKELGYAAVDDETTQQTMRELKELGYTSEPHAAVAYRA
LRDQLNPGEYGLFLGTAHPAKFKESVEAILGETLDLPKELAERADLPLLSHNLPADFAAL
RKLMMNHQ
>0003
MKKMQSIVLALSLVLVAPMAAQAAEITLVPSVKLQIGDRDNRGYYWDGGHWRDHGWWKQH
YEWRGNRWHLHGPPPPPRHHKKAPHDHHGGHGPGKHHR
...
..
.
E-mail address
The URL to access the results will be sent to this address after the assignments are completed.
GENES data set
One or more species may be specified as a template data set for KO assignment.
The computation time is proportional to the size of the data set. The accuracy will be improved if closely related species of the query are included in the data set.
- Representative set (for GENES, for Eukaryotes, for Prokaryotes)
pre-selected data set of species from each taxonomic group in GENES (about 25 species)
- Manual selection
Template species may be selected using the KEGG organism code.
KO assignment
KO assignment methods may be performed based on the bi-directional best hit (BBH, default) or single-directional best hit (SBH).
The computation of the BBH-based method takes about twice as much as that of SBH-based one. However, the BBH-based method will be more accurate than SBH-based one, if the query sequences is from a complete or draft genome. If the number of query sequences is very large such as those from metagenomes, then the SBH-based method should suffice (and save time).
Limitations
Number of the query sequences are limited by the following conditions depending on the query type and selected databases.
For amino acid sequence queries: (the number of query sequences) * (the total number of sequences in selected organisms) < 135,000,000,000
For nucleotide sequence queries: (the number of query sequences) * (the total number of sequences in selected organisms) < 135,000,000,000 / 3
Example of result
Users query list
| ID | name | state | result | start (GMT) | end (GMT) | retry | |
| 1156789000 | Human | computing | 4/01 13:00 2007 | ||||
| 1156789012 | E.coli | complete | html, text | 4/02 7:04 2007 | 4/02 7:15 2007 | ||
| 1156789055 | query070403 | failed | 4/03 12:20 2007 | retry |
'Failed' means abnormal process termination by a server or network trouble. You can retry the computation with the same file and parameters.
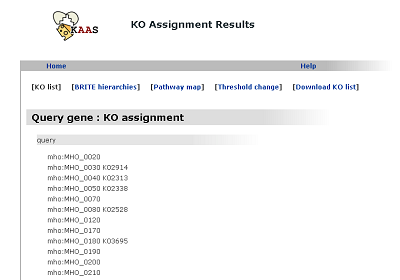
KO list
The list of query genes with the K numbers given by the KAAS.
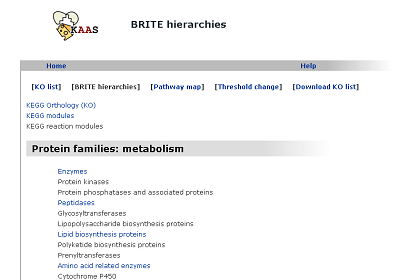
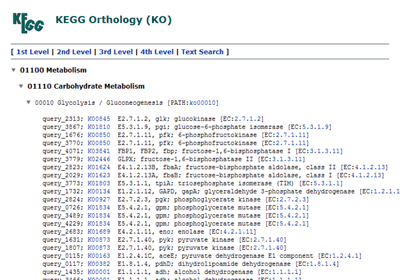
BRITE hierarchies (changed from "KO hierarchy")
The hierarchical list of annotated genes, which is categorized according to the BRITE database.
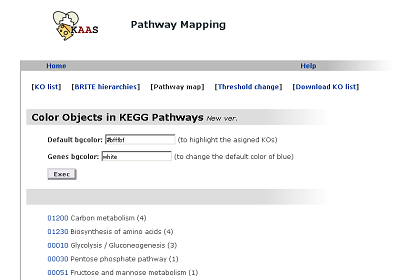
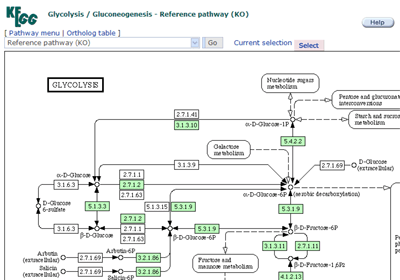
Pathway map
The list of pathways with linkes to graphical pathway maps.
Method
First, the bit scores between a query sequence and the reference sequence set (taken from the KEGG GENES database) are computed by a homology search tool, and homologs are found in the reference set. Next, homologs ranked above the threshold are selected as ortholog candidates based on the bit score and the bi-directional hit rate (BHR) defined below. Ortholog candidates are divided into KO groups according to the annotation of the KEGG GENES database. Finally, the assignment score is calculated based on the likelihood and heuristics for each KO group. Then, the K number of the KO group with the highest score is assigned to the query sequence.
Bi-directional hit rate
Given a genome to be annotated, it is compared against each genome in the reference set of the KEGG GENES database by the homology searches in both forward and reverse directions, taking each gene in genome A as a query compared against all genes in genome B, and vice versa. Those hits with bit scores less than 60 are removed. Because the bit scores of a gene pair a and b from two genomes A and B, respectively, can be different in forward and reverse directions, and because the top scores do not necessarily reflect the order of the rigorous Smith-Waterman scores, we define the BHR as:
BHR = Rf * Rr
Here, R = S'/Sb where S' is the bit score of a against b, and Sb is the score of a against the best-hit gene in genome B (which may not necessarily be b). Rf refers to the score from the forward hit (A against B), and Rr refers to the score from the reverse hit (B against A). We select those genes whose BHR is greater than 0.95 in BBH method, and Rf is greater than 0.95 in SBH method.
Assignment score
We define a score for each ortholog group in order to assign the best fitting K numbers to the query gene:
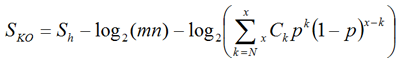
where Sh is the highest score among all ortholog candidates in the ortholog group, m and n are the sequence lengths of the query and the target of BLAST, respectively, N is the number of organisms in ortholog group, x is the number of organisms in the original ortholog group from which this group is derived, and p is the ratio of the size of the original ortholog group versus the size of the entire GENES database. The second term is for the normalization of the first term by sequence lengths, and the third term is a weighting factor to consider the number of ortholog candidates that are found in the original.
Download of stand-alone version
Stand-alone KAAS for Linux and OS X [FTP] [HTTPS]
Reference
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A., and Kanehisa, M.; KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182-W185 (2007). [pubmed] [NAR]
Feedback
Please use the feedback page to send your comments or questions to KAAS.